“Know how to solve every problem that has been solved.”“What I cannot create, I do not understand.”— Richard Feynman
Casida for Paints: Color from Conjugation
Quantum Chemistry
A carrot is orange, a tomato is red, and butadiene is colorless — and the same
equation explains all three. Color, for a molecule, means absorbing a chunk of
the visible spectrum, and a molecule absorbs a photon when it has an electronic
excitation at that photon's energy. So "what color is this compound?" is really
"where is its lowest bright excitation?", and that is a question time-dependent
density functional theory answers directly, through the Casida equations. This
page runs real TDDFT on the linear polyene series — conjugated chains of growing
length — watches the absorption march from the deep ultraviolet toward the
visible as the chain grows, hands the excitation energies to the
symbolic-regression engine
to extract the law, and tests that law against the real carotenoids — where it
captures the mechanism cleanly and misses the absolute wavelength in a way
worth understanding.
The excitation is the color
Linear-response TDDFT reduces the question of a molecule's excitations to a
single generalized eigenvalue problem, Casida's equation. Its eigenvalues are
the excitation energies ωI; its eigenvectors are the
transition densities, which set the oscillator
strengthfI — how strongly the molecule couples to light at
that energy. Solve it, find the lowest root with large f, and the
absorption wavelength follows immediately:
λmax[nm]=ω[eV]1239.84.
For a conjugated molecule the bright, low-energy transition is always the same
character: an electron promoted from the highest occupied π
orbital to the lowest unoccupied π∗ — the HOMO→LUMO
π→π∗ excitation delocalized over the conjugated
backbone. Everything about the molecule's color is set by where that one
transition sits.
# The bright pi->pi* excitation of a conjugated molecule, from real TDDFT.# Series: all-trans polyenes CH2=(CH-CH=)_{n-1}CH2, n = 1..6 double bonds.def tddft_lambda_max(n): mol = gto.M(atom=planar_polyene(n), basis='6-31g*', verbose=0) mf = dft.RKS(mol).density_fit(); mf.xc = 'b3lyp'; mf.kernel() # ground state td = tddft.TDA(mf); td.nstates = 6; td.kernel() # Casida (TDA) f = td.oscillator_strength() e = td.e * 27.2114 # eV b = int(np.argmax(f)) # brightest root = the pi->pi* 1Bu state return e[b], 1239.84 / e[b], f[b] # energy, lambda_max [nm], oscillator strength
A methods trap worth knowing: the triplet instability
There is a real subtlety here, and stepping on it is instructive. Run
full TDDFT (the Casida equations with both the excitation and
de-excitation blocks) on these polyenes and the shorter chains behave, but from
the six-carbon chain onward the calculation returns a spurious excitation at
almost zero energy with an enormous oscillator strength — a red flag, not a red
dye. The cause is a triplet instability: the closed-shell Kohn–Sham
reference for a stretched conjugated system is unstable toward a lower-energy
open-shell state, and that instability leaks into the response equations as a
near-zero root. The standard cure is the Tamm–Dancoff approximation,
which drops the de-excitation coupling block responsible for the instability.
TDA systematically blue-shifts the excitation energies by a few tenths of an eV,
but it is stable, and for polyenes it is the appropriate tool. Knowing
when your method is lying to you — and why — is the difference between running a
black box and using it.
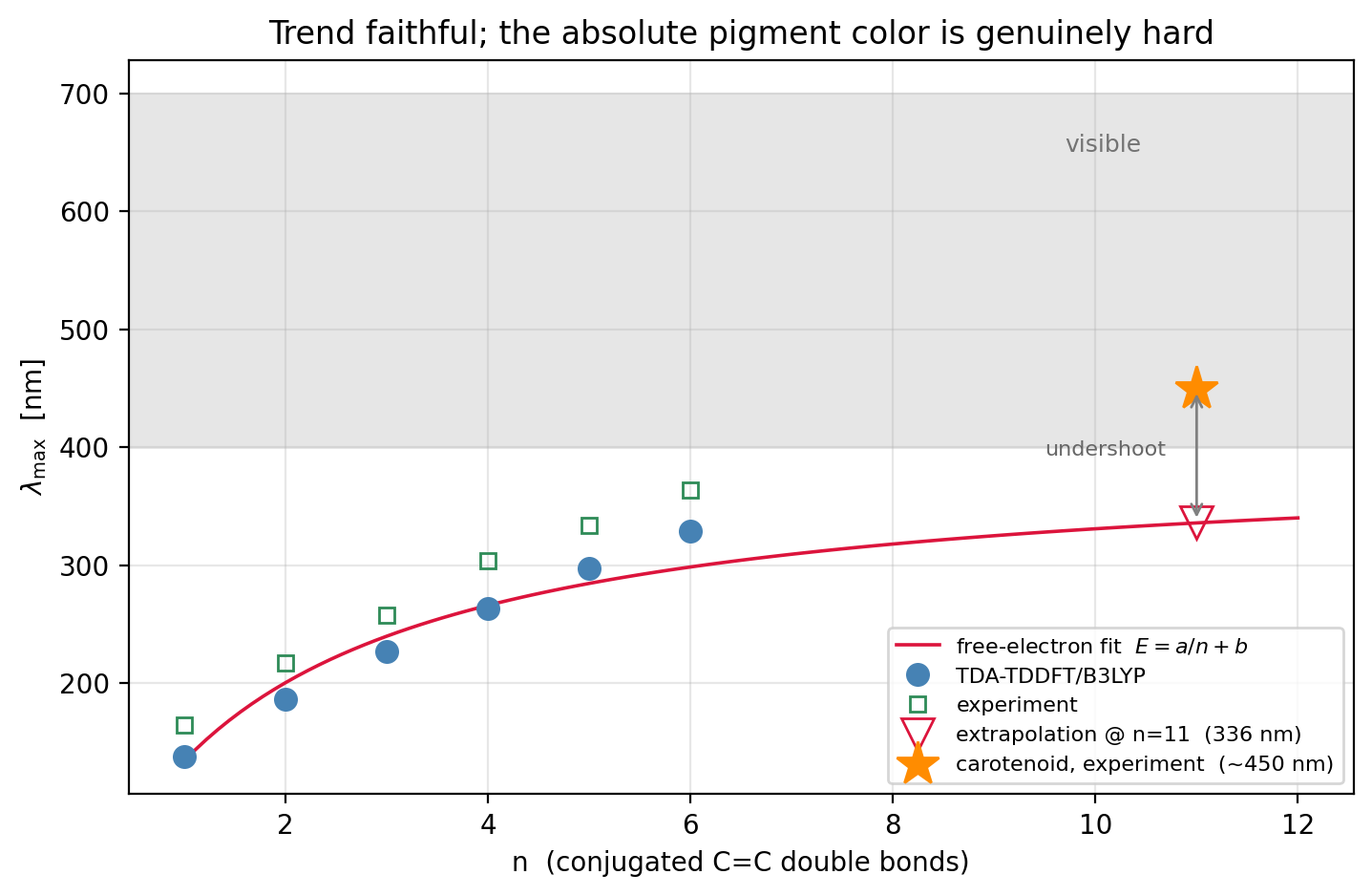
The red-shift, computed
Across the series the bright π→π∗ absorption
red-shifts steadily as the conjugated chain lengthens — the central result, and
it reproduces the experimental trend faithfully:
TDA-TDDFT / B3LYP / 6-31G* on all-trans polyenes (n = C=C double bonds) n=1 C2H4 E=8.98 eV lambda_max=138 nm exp=165 nm (-27) n=2 C4H6 E=6.65 eV lambda_max=186 nm exp=217 nm (-31) n=3 C6H8 E=5.46 eV lambda_max=227 nm exp=258 nm (-31) n=4 C8H10 E=4.71 eV lambda_max=263 nm exp=304 nm (-41) n=5 C10H12 E=4.17 eV lambda_max=297 nm exp=334 nm (-37) n=6 C12H14 E=3.77 eV lambda_max=329 nm exp=364 nm (-35) MAE vs experiment = 33 nm trend correlation r = 0.9991 law E(n): symbolic regression prefers E ~ -2.91 log(n) + 8.82 (RMSE 0.13 eV) free-electron form E = 6.09/n + 3.14 (RMSE 0.30 eV) -> log fits better in-range but -> -inf; only a/n+b has a finite gap extrapolate to carotenoids (n~11): 336 nm vs experiment ~450 nm (undershoot)
Every point sits about 30 nm short of experiment — a mean absolute error of
33 nm, and, more tellingly, a trend correlation of r=0.9991.
That near-constant offset is TDA/B3LYP's systematic blue-shift: the method
overestimates the π→π∗ energy by a few tenths of an
eV, uniformly, sliding every wavelength toward the ultraviolet while preserving
the shape of the trend almost perfectly. Color is about where the absorption
moves as the molecule changes, and that motion is reproduced to better
than a percent.
The mechanism is the particle in a box. The π electrons are
delocalized along the conjugated backbone like a free electron confined to a
wire; lengthening the chain lengthens the box, the quantized levels
Ek∝k2/L2 squeeze together, and the HOMO→LUMO gap — the
absorption energy — shrinks. More conjugation, smaller gap, redder color. The
free-electron model even
suggests the functional form, E∝1/n — though the fit has a
twist worth savoring.
Hand the six excitation energies to the symbolic-regression engine and its best
simple law is not the free-electron 1/n but a logarithm,
E≈−2.9lnn+8.8, which fits the points more than twice
as tightly (RMSE 0.13 vs 0.30 eV). Over n=1–6 the
logarithm genuinely is the better description — and it is physically wrong:
lnn→∞ means the gap collapsing without bound, an
infinite chain absorbing at infinite wavelength. The free-electron form
E=a/n+b fits worse in-range but has the one feature the physics
demands — a finite intercept b, the gap of the infinite chain. This
is the symbolic-regression
page's warning arriving on real data: the tightest fit inside your data is
not the law that extrapolates. Choosing between two equally-good-in-range fits
is where physics, not the fitter, has to speak — here through the demand that an
infinite conjugated wire still have a finite gap.
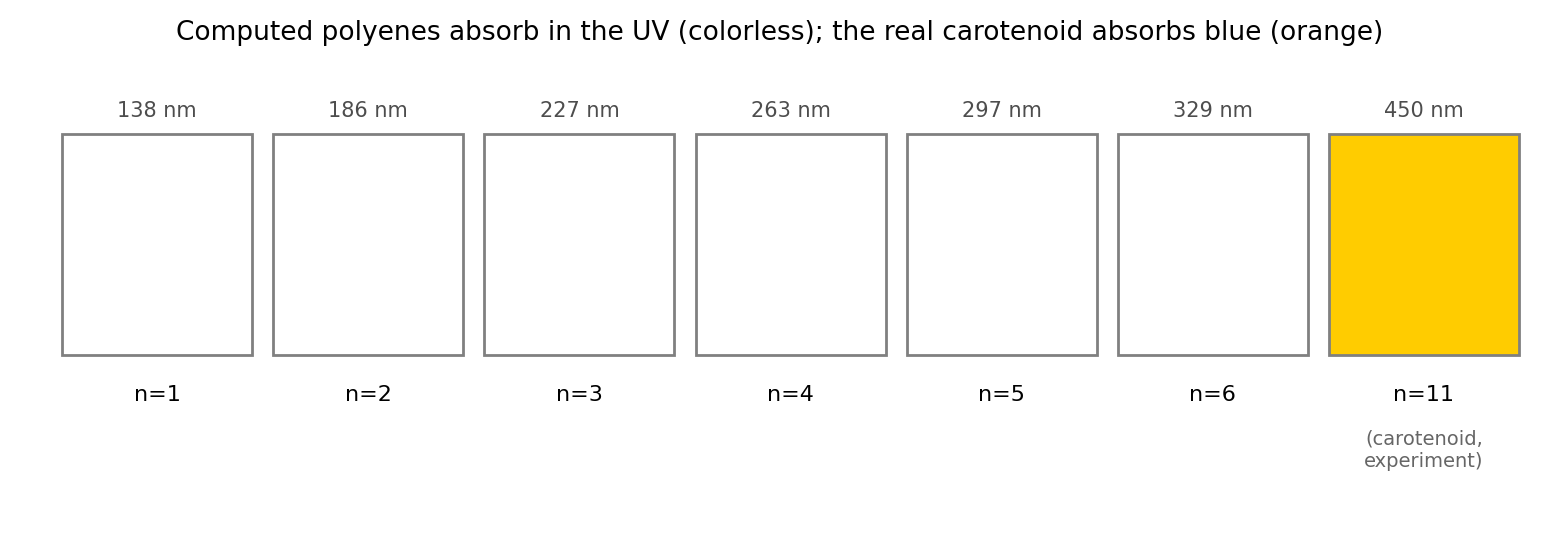
From spectrum to swatch
A pigment's perceived color is, to a first approximation, the
complement of what it absorbs: absorb blue light and the reflected
remainder looks orange; absorb nothing in the visible and the compound is
colorless. That is why the short polyenes are clear liquids — their absorption
is buried in the ultraviolet — and it is why the extrapolated carotenoid, with
its absorption pushed into the blue, comes out orange. The swatch on the right
is not a photograph; it is the computed absorption wavelength run through a
wavelength-to-color map and complemented. The physics that colors it is the same
physics that colors a carrot.
So does the law predict the orange of a carrot? Not quite — and the way it
misses is the point. Extrapolate the free-electron fit to the eleven double
bonds of β-carotene and it lands at 336 nm, still in the
near-ultraviolet, more than 100 nm short of the real ∼450 nm
that makes carotenoids orange. Three things conspire: TDA's systematic
blue-shift, an asymptotic gap the short chains cannot pin down (the fit's
3.1 eV intercept against polyacetylene's true ∼1.8 eV), and the
multireference character of long polyenes that strains any single-reference
method — the same instability that forced TDA in the first place. The mechanism
is unambiguous and the trend is quantitative; the absolute color of a real
pigment, extrapolated across a factor of two in chain length, is genuinely
hard. That gap between a clean trend and a stubborn absolute number is what
computational pigment design actually looks like.
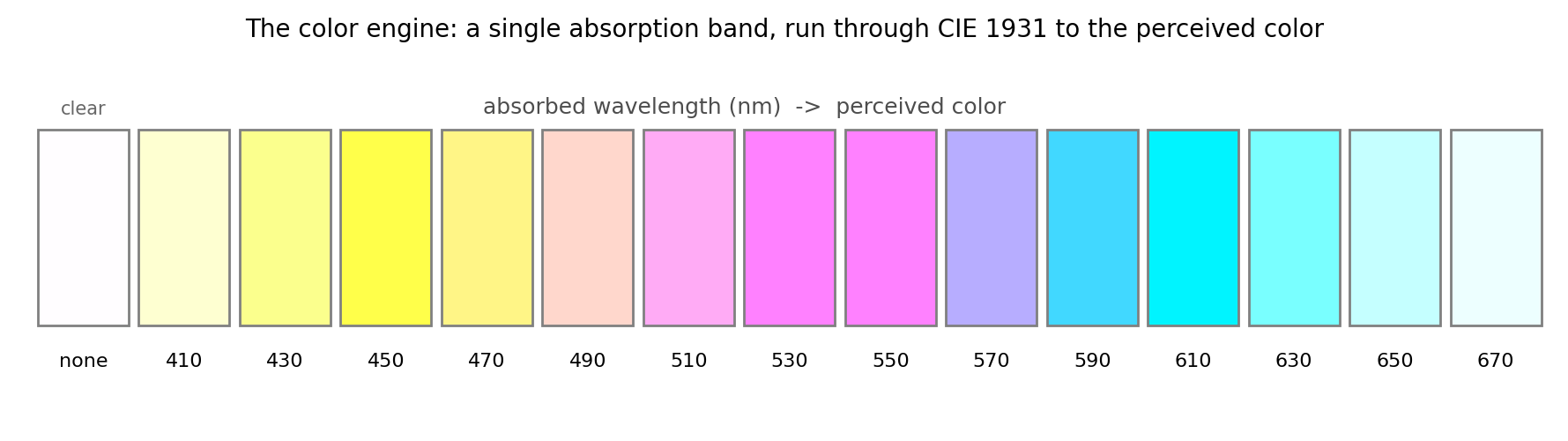
Beyond λmax: the real color engine
The complement rule is a cartoon — a good one, but a cartoon. A real pigment's
color is not set by a single wavelength; it is set by the whole absorption
band, seen through the eye. Turning a computed spectrum into an actual color is
a deterministic pipeline: broaden the TDDFT excitations into an absorption
curve, attenuate daylight through it by Beer–Lambert, and convolve what is left
with the CIE 1931
color-matching functions — the tabulated response of the three cone types —
to land in the sRGB a screen can display. Sweep a single synthetic absorption
band across the visible and the complements fall out exactly:
This is the piece the swatch above only gestured at, and it works for any
molecule, not just polyenes. Which sharpens where the difficulty actually is,
because predicting a molecule's color factors cleanly in two:
The first factor is the pipeline above — deterministic, validated, done. The
second is where the difficulty lives, and cheap TDDFT is not yet good enough to
carry it for an arbitrary dye. Screen a handful of small molecules and the
pattern is stark: the colorless controls come out right — naphthalene and
anthracene absorb only in the ultraviolet, so the engine returns white — but
indigo, a deep blue whose absorption sits at 605 nm, comes back from B3LYP with
its lowest band near 400 nm, predicting yellow. A one-electron-volt
error in the excitation energy flips the hue to its opposite. Optimizing the
geometry and adding an implicit solvent each nudge the number the right way,
but neither closes a gap that large.
So the shape of computational pigment design is this: the colorimetry is easy
and the quantum chemistry is hard. The color engine
(scripts/gen_dye_color.py) is a solved, reusable tool; the
bottleneck — the thing worth methodological effort — is getting an excitation
energy right to a tenth of an electron-volt for a molecule no one has yet
measured. That is where the field's method development lives, and it is why a
screening pipeline that generates candidate colorants is only ever as
good as the excited-state method underneath it.
What this does and doesn't get right
The trend is robust; the absolute numbers are approximate. TDA/B3LYP blue-shifts the polyene π→π∗ energies systematically, so every computed λmax sits short of experiment by a roughly constant offset. The red-shift with chain length — the thing that sets color — is reproduced faithfully.
Real pigments are not gas-phase polyenes. Solvent shifts, vibronic structure (the absorption is a band, not a line), aggregation, and the substituents of an actual dye all move the number. A quantitative color needs the full spectrum convolved with the eye's response, not a single λmax.
Long conjugation is where TDDFT strains. The same triplet instability that forced TDA is a symptom of the multireference character that grows with chain length; for the true carotenoids a single-reference method is being pushed near its limit, which is why we extrapolate a law from the tractable short chains rather than compute β-carotene directly.
Try First
Each prompt asks a checkable question about the working code or math
above — predict an output, derive a sign, state an invariant, find a
bug. Commit to an answer before clicking "reveal." That commitment is
the whole point: if your answer matched, you understand the piece you
were looking at; if it didn't, that's the part worth re-reading.
predict
The absorption red-shifts as the conjugated chain grows, following roughly
E≈a/n+b for the excitation energy. As
n→∞ (an infinitely long conjugated wire, i.e.
polyacetylene), what does this predict for the gap — and is it right?
answer
The fit predicts a finite gap: E→b as
n→∞, the nonzero intercept. The naive free-electron
model, by contrast, predicts the gap vanishing as 1/n→0 —
an infinite conjugated chain would be a gapless metal. Experiment sides with
the finite gap: polyacetylene is a semiconductor with a gap around
1.5–2 eV, not a metal. The reason is Peierls distortion — the
chain lowers its energy by alternating short (double) and long (single)
bonds, and that bond-length alternation opens a gap at the Fermi level
exactly as a periodic potential opens a band gap. It is why real polyenes
have alternating bonds at all, and why λmaxsaturates for long chains instead of running off to infinity. A
pigment can only get so red by adding conjugation.
why does this work
Ground-state DFT already gives you a HOMO and a LUMO. Why not just call the
HOMO–LUMO orbital energy gap the absorption energy and skip TDDFT entirely?
answer
Because the HOMO–LUMO gap of Kohn–Sham orbitals is not an excitation energy
— it is a difference of auxiliary eigenvalues with no rigorous claim to be
the energy of a real excited state, and for most functionals it is off by
electron-volts. An electronic excitation is a genuinely many-body event: the
promoted electron and the hole it leaves behind attract each other, other
electrons relax around them, and the true transition energy is the
HOMO–LUMO gap corrected by all of that. TDDFT's job is precisely to add
those corrections — the response of the density to the perturbing field
builds in the electron–hole interaction through the exchange–correlation
kernel. That is what Casida's equation computes, and it is why the bright
state comes out at a physically meaningful energy while the bare orbital gap
does not. Skipping TDDFT would get the trend roughly and the number wrong.
Reproduce it
scripts/gen_casida_paints.py builds the planar all-trans polyenes
from scratch (bond-alternating geometry, no conformer lottery), runs
B3LYP + TDA in PySCF, and extracts the bright π→π∗
state for n=1 to 6. It then imports the
genetic-programming engine from the symbolic-regression
page — the same code that rediscovered Kepler's law — to recover
E(n) from the computed points, and maps each
λmax to a color swatch. Change the functional, the
basis, or the series and watch the trend hold and the offset move.