Project: Self-learning Monte Carlo on a PySCF surface
Projects
The catch with doing statistical mechanics on quantum-chemical energies is the cost: every Monte Carlo step wants the energy of a new configuration, and a PySCF calculation per step is unaffordable once the conformational space is large. The Wang-Landau project dodged this — butane was a precomputed scan, the chain used a hand-built cheap energy. Self-learning Monte Carlo (SLMC) is the principled way not to dodge it: learn a cheap surrogate energy from a modest set of PySCF calculations, use it to propose moves, and accept against the true PySCF energy. The surrogate makes the sampling affordable; the true-energy accept/reject keeps it exact.
The distinction that makes this worth doing: a plain machine-learned potential replaces PySCF and inherits whatever error it has — bias you can't see and can't bound. SLMC keeps PySCF as the truth and lets the surrogate be a guess that gets corrected. Propose a move drawn from the surrogate's Boltzmann distribution, then accept with
The acceptance depends only on the difference between true and effective energies. If the surrogate tracks PySCF well, that difference barely changes from step to step, acceptance stays high, and the chain decorrelates fast — but detailed balance is enforced with respect to the true distribution, so the stationary distribution is the exact PySCF one no matter how crude the surrogate. The one PySCF call per step is the price; the payoff is that the surrogate's proposals are good enough to need few such steps.
# SLMC: propose from a cheap learned surrogate, accept against true PySCF.
# Samples P_true ∝ exp(-β E_true) EXACTLY, even when the surrogate is wrong —
# the surrogate only affects efficiency, never the answer.
def slmc_step(state, beta, E_true, E_eff, propose_from_surrogate, rng):
prop = propose_from_surrogate() # cheap: NO PySCF here
# acceptance depends only on the change in (true - effective) energy
d = (E_true(prop) - E_eff(prop)) \
- (E_true(state) - E_eff(state))
if np.log(rng.random()) <= -beta * d: # the single PySCF call: E_true(prop)
return prop # accepted
return state # rejectedA test with a coupling the surrogate can't see
Pentane has two central dihedrals, and , and they are coupled: when both go gauche of opposite sign (g⁺g⁻), the two terminal methyl groups collide. This is the textbook syn-pentane interaction, and it's exactly the kind of thing an additive (per-dihedral) surrogate cannot represent. A rigid PySCF scan of the 2D surface shows it plainly — anti-anti is the global minimum, single-gauche costs ~2 kcal/mol, g⁺g⁺ about 3.5, but the g⁺g⁻ corner spikes to ~50 kcal/mol (a rigid-scan exaggeration of the real ~3 kcal/mol penalty — which makes the surrogate's failure all the more visible).
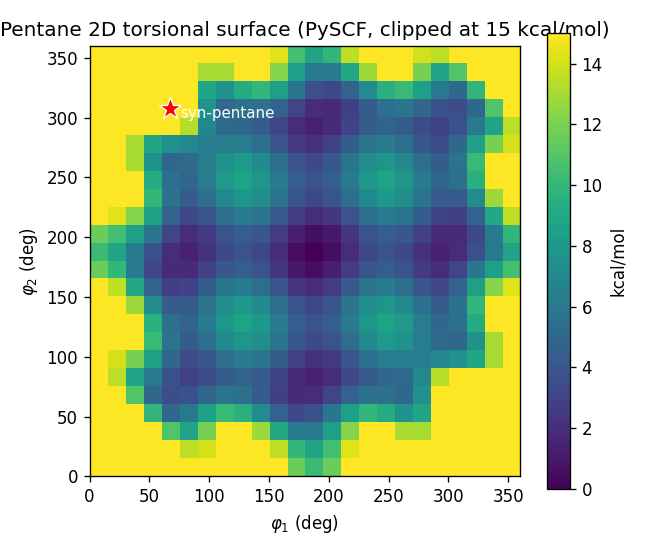
The surrogate is built to fail in a specific, instructive way: take the single-dihedral torsional profile (the slice of the surface with the other dihedral held anti) and assume the two dihedrals are independent. That's a handful of PySCF points, not the full grid — and it predicts g⁺g⁻ ≈ g⁺g⁺ ≈ 4 kcal/mol, missing the 50 kcal/mol clash entirely.
# The (deliberately biased) additive surrogate: each dihedral's torsional
# profile, with NO coupling between them. Trained on the 1D slice E(phi, anti)
# — 2K PySCF points, not the full K-dimensional surface.
f = E_grid[:, anti_index] # single-dihedral torsional profile
E_eff = lambda i, j: f[i] + f[j] # predicts g+g- ≈ g+g+ (misses syn-pentane)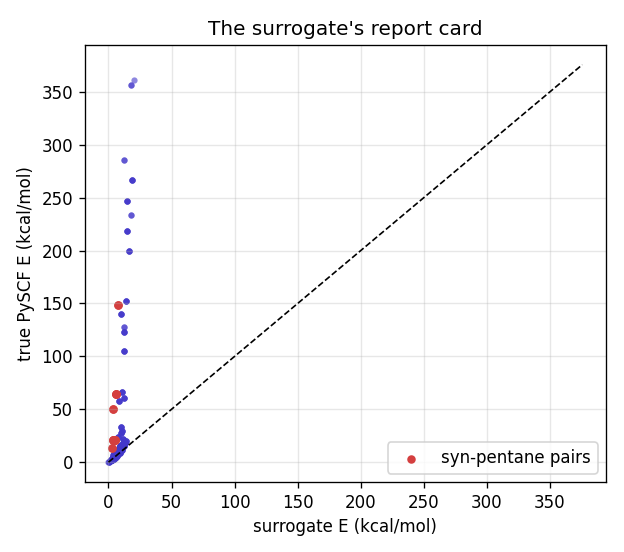
The scatter is the surrogate's report card. Most configurations sit near the diagonal (the additive model is fine there), but the syn-pentane points fly off it: the surrogate says ~4 kcal/mol, the truth says up to 50. A sampler that trusted this surrogate would happily populate states that are physically forbidden.
The demonstration: exact populations from a biased surrogate
Three ways to get the conformer populations at 600 K. Truth: a full Boltzmann sum over the 24×24 PySCF grid (576 calculations — the expensive ground truth). Surrogate only: sample the additive surrogate's Boltzmann distribution, never touching PySCF again — this is what a plain learned potential gives you. SLMC: propose from the surrogate, accept against true PySCF.
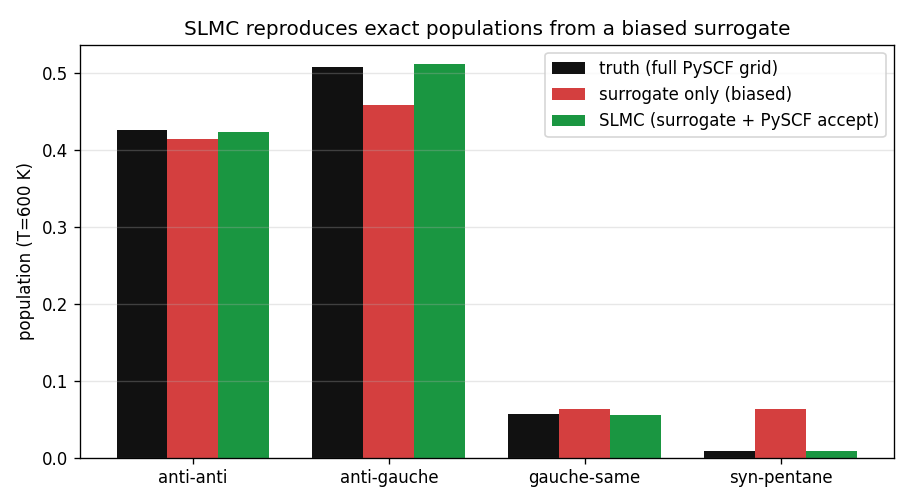
The result, in one number: the surrogate-only run puts 6.3% of the population in the syn-pentane basin; the truth is 0.9%. The additive surrogate overpopulates the forbidden conformer 7-fold, because it can't see the methyl clash. SLMC, proposing from that same biased surrogate but correcting with the true PySCF energy, gives 0.9% — matching the exact grid populations to within 0.004 in every basin, at 83% acceptance. The surrogate's blindness costs efficiency, not accuracy.
That is the entire case for SLMC over a learned potential. Both use a cheap model. The learned potential reports the surrogate's answer (6.3% — wrong). SLMC reports the truth (0.9%), using the surrogate only to decide where to look. The accept/reject against PySCF is what converts a fast-but-biased model into a fast-and-exact sampler.
Where the speedup is — and where it isn't
On this 576-point grid the SLMC run queried 500 unique PySCF energies, so the call savings are modest — the grid is small enough to nearly enumerate. That's the limit of a 2-dihedral demo: the dramatic win is exactness, and the efficiency win only becomes large where you can't enumerate at all. The mechanism is decorrelation: a good surrogate proposes big, physically-sensible jumps that a naive single-dihedral Metropolis can't, so each independent sample costs far fewer PySCF evaluations. Push to a molecule with a dozen dihedrals ( rotamer states) at a correlated level of theory where a single energy is seconds to minutes, and the difference between "PySCF every step" and "PySCF only at accept/reject, with a surrogate that lands 80% of its proposals" is the difference between intractable and routine.
The arc this completes
Three pieces, one pipeline for quantum-accurate statistical mechanics:
- PySCF — the true energy, grounded in electronic structure rather than a fitted potential.
- Wang-Landau — energies into all-temperature thermodynamics, via a density of states — the count of configurations at each energy — from one run.
- SLMC — how to afford the true energy inside the sampling loop: a learned surrogate proposes, PySCF corrects, exactness intact.
The natural fusion is self-learning Wang-Landau: the surrogate proposes moves, the flat-histogram bias drives the random walk through energy, and PySCF gates acceptance — giving the ab-initio density of states of a flexible molecule that brute force could never reach. The surrogate could itself be refined on the fly from the PySCF energies the run generates, closing the "self-learning" loop.
Falsifiable claim, and limitations
The claim: SLMC samples the exact PySCF Boltzmann distribution regardless of the surrogate's bias, because the accept/reject is against the true energy. The experiment is the populations bar chart — the additive surrogate is wrong by 7× on syn-pentane, and SLMC still reproduces the exact grid populations. It would be falsified by a converged SLMC run whose populations disagreed with the full-grid Boltzmann sum; run correctly, with the difference-of-energies acceptance, it does not.
- The PySCF surface is a rigid RHF/STO-3G scan, which exaggerates the syn-pentane penalty (~50 vs the real ~3 kcal/mol). This makes the surrogate's failure vivid but is not a quantitative pentane prediction.
- The efficiency win is asserted by scaling argument, not measured here — the 2-dihedral grid is too small for SLMC to beat enumeration on call count. The exactness win is fully demonstrated; the call-count win needs a genuinely high-dimensional system to show.
- The surrogate here is fixed (trained once on the anti-slice). A production SLMC would retrain it adaptively on the accumulating PySCF data — the actual "self-learning" loop, which this prototype stops short of.
Related on this site
- Self-Learning Monte Carlo — the method in its original lattice setting.
- Wang-Landau on a PySCF surface — the thermodynamics half of this pipeline.
- MCMC from scratch — the fixed-temperature sampling SLMC accelerates.
- I will calculate what is computable — the validate-then-extrapolate stance: the surrogate is a guess, PySCF is the truth.