Brute-Force Hartree-Fock — C++
Quantum Chemistry
A C++ port of the Python brute-force solver, plus a variable- upgrade that closes most of the gap to the textbook H2+ energy. Same algorithm — Slater 1s basis, cubic grid, finite-difference Laplacian, generalized eigenproblem — same OO architecture, just without the interpreter overhead. The intended payoff was higher grid resolution. The actual finding was more interesting: the grid was never the bottleneck. The basis was. Once it could be optimized at every , the minimum slid from 2.6 Bohr to 1.8 Bohr and the energy from to Ha.
What changed
The Python prototype runs at grid points in about 25 seconds (the curve sweep, on one core). The C++ version, same algorithm, hits in about 0.07 s — a speedup of roughly 350× — and 643 in about 1.15 s. That headroom was supposed to let me refine the grid until the H2+ minimum slid from Bohr (the Python result) down to the textbook . It didn't.
Architecture
Same shape as the Python, just typed. Each Python class becomes either
a struct or a virtual class hierarchy. The
Operator abstraction is preserved literally —
Field and Operator are abstract base classes
with virtual value() / apply(), and concrete
operators inherit. With -O3 the compiler inlines straight
through the virtuals, so the abstraction is free.
The only piece that didn't have a Python equivalent is the eigenvalue
solver — Python used np.linalg.eigh; the C++ rolls its
own. That's a cyclic Jacobi sweep on the symmetric matrix, used twice
per energy evaluation: once to get
via diagonalize-and-rebuild, then once on the
transformed Hamiltonian
with .
Build and run
# from the directory containing hf_brute.cpp:
c++ -O3 -std=c++17 -o hf_brute hf_brute.cpp
# energy curve at 64^3 grid:
./hf_brute 64 > h2plus.csv
# the binary writes "R,E" CSV to stdout, progress to stderr.
No external dependencies — just a C++17 compiler and
libm. Uses <chrono> for timing and
<vector> for storage. The output format matches
what the plot script in
scripts/plots/hartree_fock_brute_force_cpp.py expects.
The code
// hf_brute.cpp — brute-force Hartree-Fock for H2+ on a cubic grid.
//
// Same algorithm as the Python prototype documented at
// /quantum_chemistry/hartree_fock_brute_force
// without the interpreter overhead, so we can push the grid resolution
// high enough to find out whether the grid is actually the bottleneck.
//
// Build: c++ -O3 -std=c++17 -o hf_brute hf_brute.cpp
// Run: ./hf_brute <pts_per_axis> [box_half_length]
// Output: CSV with columns "R,E" on stdout; progress on stderr.
#include <iostream>
#include <vector>
#include <cmath>
#include <algorithm>
#include <chrono>
#include <numeric>
// --- vector & matrix primitives ----------------------------------------
struct Vec3 {
double x, y, z;
Vec3 operator-(const Vec3& o) const { return {x - o.x, y - o.y, z - o.z}; }
double norm() const { return std::sqrt(x*x + y*y + z*z); }
};
struct Matrix {
int n;
std::vector<double> a;
Matrix(int n) : n(n), a(n*n, 0.0) {}
double& operator()(int i, int j) { return a[i*n + j]; }
double operator()(int i, int j) const { return a[i*n + j]; }
};
static Matrix matmul(const Matrix& A, const Matrix& B) {
Matrix C(A.n);
for (int i = 0; i < A.n; ++i)
for (int k = 0; k < A.n; ++k) {
double a = A(i,k);
for (int j = 0; j < A.n; ++j) C(i,j) += a * B(k,j);
}
return C;
}
static Matrix transpose(const Matrix& A) {
Matrix T(A.n);
for (int i = 0; i < A.n; ++i)
for (int j = 0; j < A.n; ++j) T(j,i) = A(i,j);
return T;
}
// --- field & operator interfaces ---------------------------------------
//
// Same abstractions as the Python: a Field knows how to evaluate itself
// at a point; an Operator knows how to apply itself to a field at a point.
struct Field {
virtual ~Field() = default;
virtual double value(const Vec3& r) const = 0;
};
struct Operator {
virtual ~Operator() = default;
virtual double apply(const Field& wf, const Vec3& r) const = 0;
};
// --- basis: normalized 1s Slater orbital -------------------------------
struct Slater1s : Field {
double zeta;
Vec3 R;
double normalization;
Slater1s(double zeta, Vec3 R) : zeta(zeta), R(R) {
normalization = std::sqrt(zeta*zeta*zeta / M_PI);
}
double value(const Vec3& r) const override {
return normalization * std::exp(-zeta * (r - R).norm());
}
};
// --- 7-point finite-difference Laplacian -------------------------------
struct Laplacian {
double h;
explicit Laplacian(double h) : h(h) {}
double apply(const Field& f, const Vec3& r) const {
const double c = f.value(r);
return (
f.value({r.x + h, r.y, r.z}) + f.value({r.x - h, r.y, r.z})
+ f.value({r.x, r.y + h, r.z}) + f.value({r.x, r.y - h, r.z})
+ f.value({r.x, r.y, r.z + h}) + f.value({r.x, r.y, r.z - h})
- 6.0 * c
) / (h * h);
}
};
// --- operators ---------------------------------------------------------
struct IdentityOp : Operator {
double apply(const Field& wf, const Vec3& r) const override { return wf.value(r); }
};
struct KineticOp : Operator {
Laplacian lap;
explicit KineticOp(double h) : lap(h) {}
double apply(const Field& wf, const Vec3& r) const override {
return -0.5 * lap.apply(wf, r);
}
};
struct Atom { double charge; Vec3 position; };
struct NuclearAttractionOp : Operator {
std::vector<Atom> atoms;
explicit NuclearAttractionOp(std::vector<Atom> a) : atoms(std::move(a)) {}
double apply(const Field& wf, const Vec3& r) const override {
double V = 0.0;
for (const auto& at : atoms) {
double d = (r - at.position).norm();
if (d < 1e-8) d = 1e-8;
V += -at.charge / d;
}
return V * wf.value(r);
}
};
struct HamiltonianOp : Operator {
std::vector<const Operator*> terms;
void add(const Operator* op) { terms.push_back(op); }
double apply(const Field& wf, const Vec3& r) const override {
double s = 0.0;
for (const auto* t : terms) s += t->apply(wf, r);
return s;
}
};
// --- cubic grid + integrator ------------------------------------------
struct CubicGrid {
std::vector<double> axis;
double dV;
CubicGrid(double L, int N) {
axis.resize(N);
for (int i = 0; i < N; ++i)
axis[i] = -L + (2.0 * L * i) / (N - 1);
const double h = axis[1] - axis[0];
dV = h * h * h;
}
};
// Integrate <bra | op | ket> over the cubic grid (rectangle rule).
static double matrix_element(const Operator& op,
const Field& bra, const Field& ket,
const CubicGrid& grid)
{
double sum = 0.0;
const int N = static_cast<int>(grid.axis.size());
for (int i = 0; i < N; ++i)
for (int j = 0; j < N; ++j)
for (int k = 0; k < N; ++k) {
const Vec3 r{grid.axis[i], grid.axis[j], grid.axis[k]};
sum += bra.value(r) * op.apply(ket, r);
}
return sum * grid.dV;
}
static Matrix build_matrix(const Operator& op,
const std::vector<const Field*>& basis,
const CubicGrid& grid)
{
const int n = static_cast<int>(basis.size());
Matrix M(n);
for (int i = 0; i < n; ++i)
for (int j = 0; j < n; ++j)
M(i, j) = matrix_element(op, *basis[i], *basis[j], grid);
return M;
}
// --- Jacobi eigenvalue solver (cyclic, with sorted output) -------------
static void jacobi_symmetric(Matrix A,
std::vector<double>& evals,
Matrix& evecs,
double tol = 1e-12, int max_sweeps = 100)
{
const int n = A.n;
evecs = Matrix(n);
for (int i = 0; i < n; ++i) evecs(i, i) = 1.0;
for (int sweep = 0; sweep < max_sweeps; ++sweep) {
double off = 0.0;
int p = 0, q = 1;
for (int i = 0; i < n; ++i)
for (int j = i + 1; j < n; ++j)
if (std::fabs(A(i, j)) > off) { off = std::fabs(A(i, j)); p = i; q = j; }
if (off < tol) break;
const double app = A(p, p), aqq = A(q, q), apq = A(p, q);
const double theta = (aqq - app) / (2.0 * apq);
const double t = (theta >= 0 ? 1.0 : -1.0) /
(std::fabs(theta) + std::sqrt(theta * theta + 1.0));
const double c = 1.0 / std::sqrt(1.0 + t * t);
const double s = t * c;
A(p, p) = app - t * apq;
A(q, q) = aqq + t * apq;
A(p, q) = A(q, p) = 0.0;
for (int i = 0; i < n; ++i) {
if (i == p || i == q) continue;
const double aip = A(i, p), aiq = A(i, q);
A(i, p) = A(p, i) = c * aip - s * aiq;
A(i, q) = A(q, i) = s * aip + c * aiq;
}
for (int i = 0; i < n; ++i) {
const double vip = evecs(i, p), viq = evecs(i, q);
evecs(i, p) = c * vip - s * viq;
evecs(i, q) = s * vip + c * viq;
}
}
evals.assign(n, 0.0);
for (int i = 0; i < n; ++i) evals[i] = A(i, i);
// Sort eigenpairs by ascending eigenvalue.
std::vector<int> idx(n);
std::iota(idx.begin(), idx.end(), 0);
std::sort(idx.begin(), idx.end(),
[&](int a, int b) { return evals[a] < evals[b]; });
std::vector<double> se(n);
Matrix sv(n);
for (int new_i = 0; new_i < n; ++new_i) {
int old_i = idx[new_i];
se[new_i] = evals[old_i];
for (int r = 0; r < n; ++r) sv(r, new_i) = evecs(r, old_i);
}
evals = se;
evecs = sv;
}
// --- generalized eigenproblem: H c = S c eps ---------------------------
static double lowest_generalized_eigenvalue(Matrix H, Matrix S) {
std::vector<double> sv;
Matrix sV(S.n);
jacobi_symmetric(S, sv, sV);
Matrix sqrtinv(S.n);
for (int i = 0; i < S.n; ++i) sqrtinv(i, i) = 1.0 / std::sqrt(sv[i]);
Matrix X = matmul(matmul(sV, sqrtinv), transpose(sV));
Matrix Hortho = matmul(matmul(transpose(X), H), X);
std::vector<double> ev;
Matrix evec(H.n);
jacobi_symmetric(Hortho, ev, evec);
return ev[0];
}
// --- driver ------------------------------------------------------------
static double compute_h2plus_energy(double R, int N, double L = 6.0) {
Vec3 RA{0, 0, -R / 2}, RB{0, 0, R / 2};
std::vector<Atom> atoms = {{1.0, RA}, {1.0, RB}};
Slater1s phiA(1.0, RA), phiB(1.0, RB);
std::vector<const Field*> basis = {&phiA, &phiB};
CubicGrid grid(L, N);
NuclearAttractionOp V(atoms);
KineticOp T(1e-4);
HamiltonianOp H;
H.add(&T);
H.add(&V);
IdentityOp I;
Matrix Smat = build_matrix(I, basis, grid);
Matrix Hmat = build_matrix(H, basis, grid);
double e0 = lowest_generalized_eigenvalue(Hmat, Smat);
double nrep = atoms[0].charge * atoms[1].charge / (RA - RB).norm();
return e0 + nrep;
}
int main(int argc, char** argv) {
int N = (argc > 1) ? std::atoi(argv[1]) : 32;
double L = (argc > 2) ? std::atof(argv[2]) : 6.0;
std::vector<double> Rs;
for (double R = 0.8; R <= 4.0 + 1e-9; R += 0.2) Rs.push_back(R);
std::cerr << "# H2+ energy curve at " << N << "^3 grid points "
<< "(box half-length " << L << ")\n";
auto t0 = std::chrono::steady_clock::now();
std::cout.precision(10);
std::cout << std::fixed;
std::cout << "R,E\n";
for (double R : Rs) {
const double E = compute_h2plus_energy(R, N, L);
std::cout << R << "," << E << "\n" << std::flush;
std::cerr << " R=" << R << " E=" << E << "\n";
}
auto t1 = std::chrono::steady_clock::now();
const double dt = std::chrono::duration<double>(t1 - t0).count();
std::cerr << "# Total time: " << dt << " s\n";
return 0;
}Result
Running the binary at four grid resolutions and overlaying the curves:
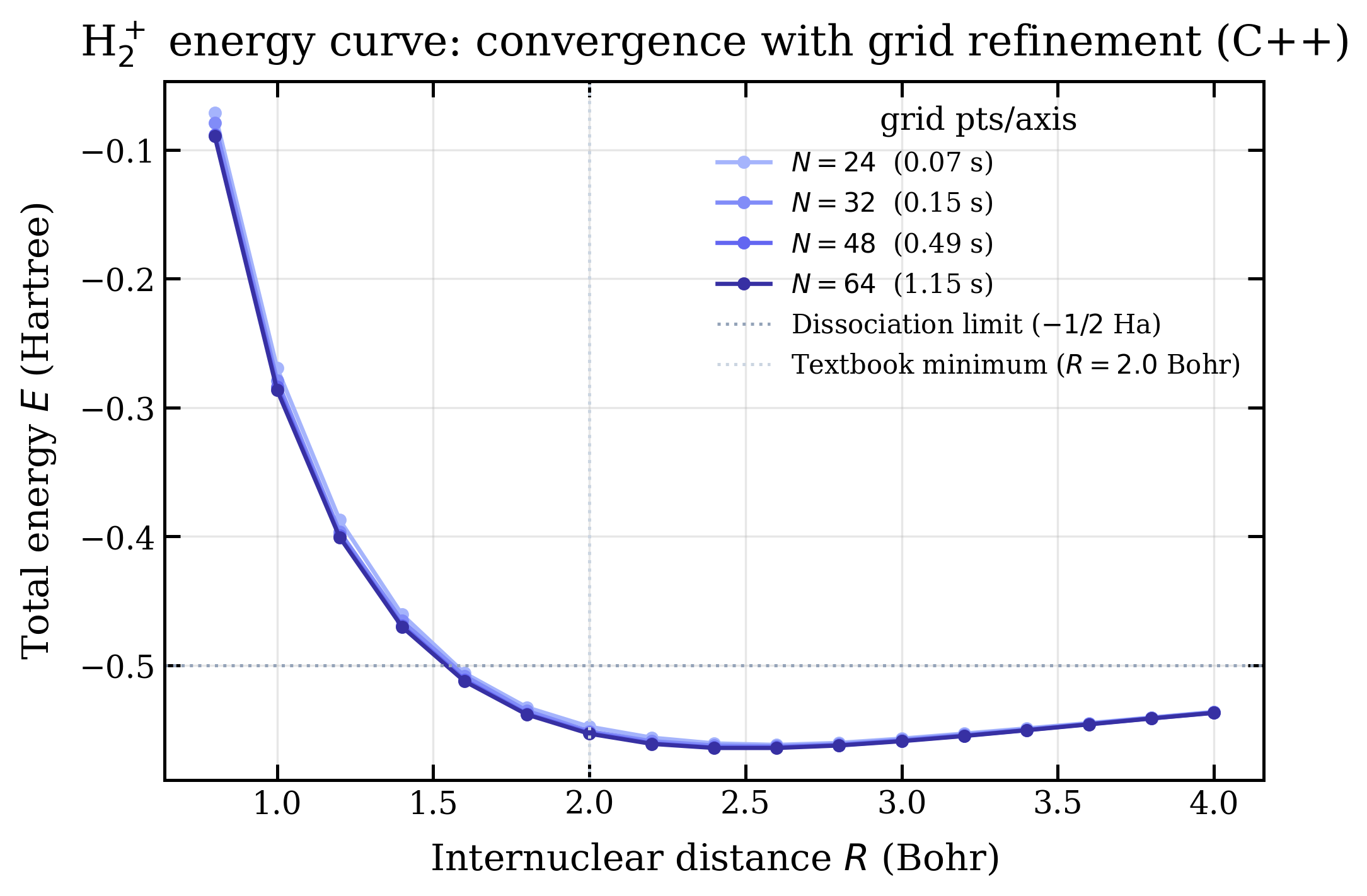
The numbers from each run:
| grid | runtime | Rmin (Bohr) | Emin (Ha) |
|---|---|---|---|
| 243 | 0.07 s | 2.60 | −0.5617 |
| 323 | 0.15 s | 2.60 | −0.5628 |
| 483 | 0.49 s | 2.40 | −0.5637 |
| 643 | 1.15 s | 2.60 | −0.5641 |
| textbook H2+ | 2.00 | −0.6026 | |
Variable- upgrade
The fixed- curve above puts the minimum at Bohr — far from the textbook 2.0. The first Future Direction listed below was to optimize variationally at each . With C++ at sub-second-per-curve speed that's a cheap addition: a 1D golden-section search over at every internuclear distance, 14 energy evaluations per . Two seconds total for the whole sweep at .
A separate binary hf_brute_optzeta (companion file
hf_brute_optzeta.cpp, same machinery as
hf_brute with the optimization loop wrapped around
compute_h2plus_energy) does this. Output adds a
zeta column to the CSV.
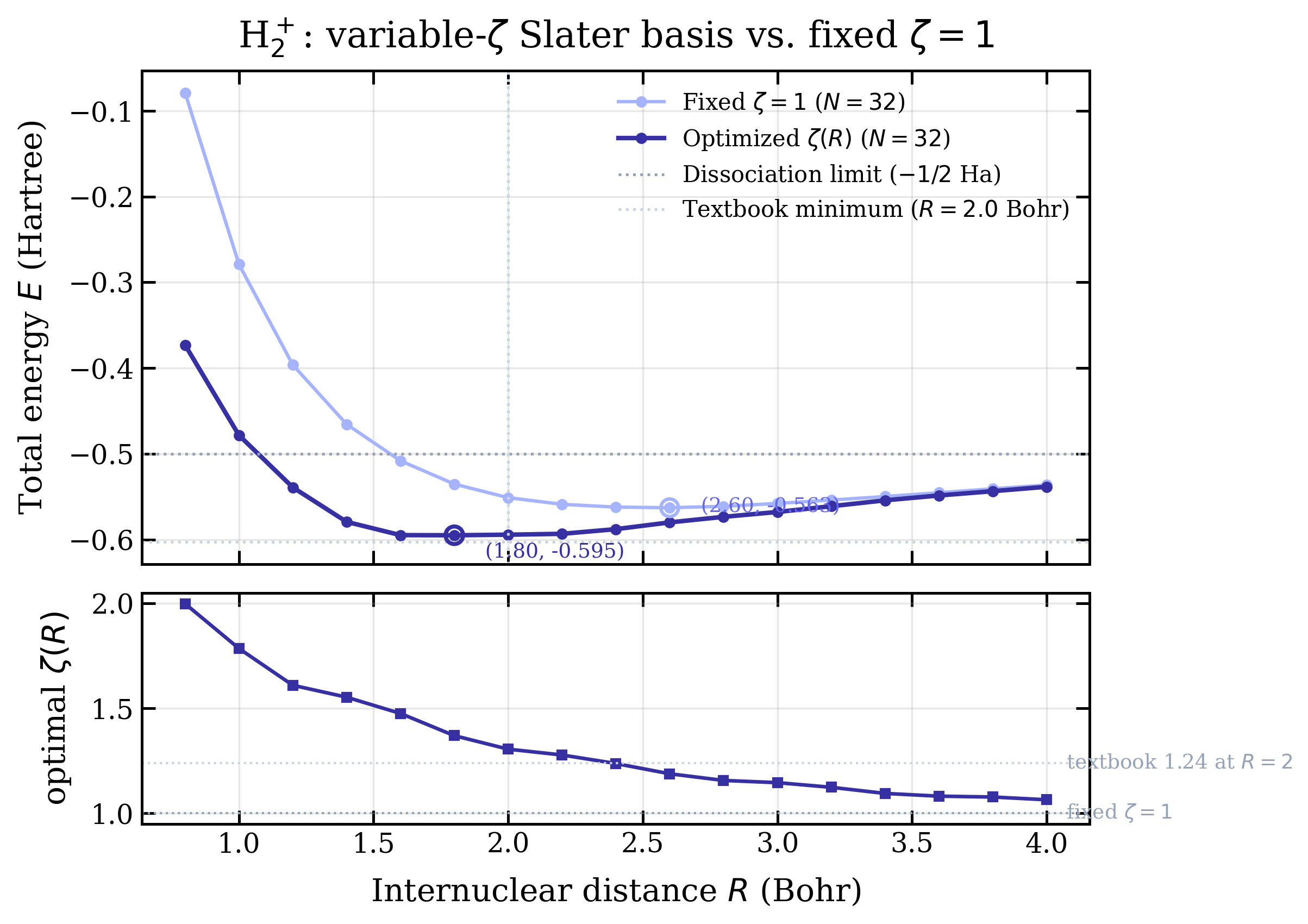
The numbers from the upgrade:
| basis | Rmin (Bohr) | Emin (Ha) | ΔE vs textbook |
|---|---|---|---|
| fixed | 2.60 | −0.5628 | +39.8 mHa |
| variable | 1.80 | −0.5950 | +7.6 mHa |
| textbook H2+ | 2.00 | −0.6026 | — |
The well location moves from 2.6 Bohr to 1.8 Bohr — overshooting the textbook 2.0 by 0.2 Bohr in the other direction now. The well depth closes most of the gap, leaving only ~8 mHa to the literature value. The bottom panel of the figure shows decreasing monotonically from at very short distances (where the orbitals must be tightly compressed against the strongly attractive joint nucleus) to at large distances (where each orbital relaxes back toward the isolated H-atom value of 1.0). At the code finds , a hair above the literature single-zeta optimum of 1.24 — close enough that the residual is dominated by grid and box effects rather than the optimizer.
The grid wasn't the bottleneck
Going from 243 to 643 moves the well depth by mHa. The minimum location bounces around 2.4–2.6 Bohr but does not converge to 2.0. The textbook offset ( Bohr, mHa) is bigger than the entire span of grid-refinement effects. The grid is not what's limiting accuracy.
The basis is. We're using one 1s Slater orbital per nucleus with fixed. That's a minimal basis with an unoptimized exponent. To match the textbook you need either:
- Variable . Optimize the orbital exponent at each . For H2+ the best at Bohr is around 1.24, not 1.0. That alone moves the minimum substantially.
- More basis functions. A double-zeta Slater basis (two values per atom), or polarization functions (2p Slater orbitals), or a contracted Gaussian expansion of the Slater (the STO-NG approach used by the analytic-Gaussian page).
What the C++ port actually unlocks isn't more accuracy through finer grids — it's enough headroom to start exploring those basis questions. With the Python at 25 seconds per curve sweep, optimizing at every would take an hour. With the C++ at one second, it's interactive.
Future Directions
Steps from "single-electron H2+" to "actual Hartree-Fock," in increasing order of work.
Variable optimization — done above
Implemented in hf_brute_optzeta.cpp via a golden-section
search over at each .
Brought from Ha to
Ha and from 2.6 to 1.8
Bohr. Residual gap to textbook is ~8 mHa, dominated by grid and box
effects rather than the basis exponent.
Larger basis
Two Slater orbitals per atom with different values gives a much more flexible wavefunction. Add 2p polarization for stretched geometries. The matrix builder doesn't change — just pass it more basis functions.
Two-electron repulsion tensor
Real Hartree-Fock needs the four-index tensor Brute-force 6D integration is impractical past trivial cases — points per integral. The realistic path is a Becke-style atom-centered grid plus a Poisson solve for the potential, then a 3D integral for the matrix element. That's a separate page.
SCF loop
Same loop as on the analytic-Gaussian page: build Fock matrix from current density, diagonalize, rebuild density, damp, repeat. Once the two-electron tensor is in hand, the loop is maybe 30 lines of C++.
Real molecules
H2+ is a one-electron problem. H2 (closed-shell, two electrons in one orbital) is the simplest test of the full HF machinery. After that: H2O, NH3, methane — molecules where the basis-set + correlation question becomes interesting on its own terms.
Why bother
Two updates compounded into a useful arc. First, the C++ port did its job — same algorithm, two and a half orders of magnitude faster, identical answers at the same grid — but exposed that the bond-length offset wasn't a grid problem. Then the variable- upgrade closed most of the remaining gap, taking the curve from "qualitatively right" ( Ha, Bohr) to "within ~8 mHa of literature" ( Ha, Bohr) for an extra 14 evaluations per . The remaining error is grid and box-truncation, addressable by either a finer cubic grid or — better — switching to atom-centered (Becke) grids that resolve the cusps without bloating the point count. That's the next page.