LAPW Method
Solid State Physics
What you need to know first 3 concepts, 3 layers
The requisite-knowledge inventory for this page, bottom-up: the primitives at the base, combined upward until you reach what this page assumes. Skim the layers you already own; start wherever the ground gets unfamiliar.
- base
- L1
- L2
- ↳you are here
The Linearized Augmented Plane Wave (LAPW) method is a powerful technique for solving the electronic structure problem in periodic solids. It combines plane waves in the interstitial region with atomic-like functions inside muffin-tin spheres.
Basic Idea
The LAPW method divides the unit cell into two regions:
- Muffin-tin spheres: Around each atom, where the potential is nearly spherically symmetric
- Interstitial region: The space between atoms, where the potential is relatively smooth
Different basis functions are used in each region and matched at the boundaries.
LAPW Basis Functions
The LAPW basis function is defined piecewise:
where:
- is the radial solution of the Schrödinger equation at energy
- is the energy derivative of the radial solution
- are spherical harmonics
- where are reciprocal lattice vectors
- is the muffin-tin radius for atom
Radial Schrödinger Equation
Inside the muffin-tin sphere, the radial wavefunction satisfies:
where the radial Hamiltonian is:
The energy derivative satisfies:
This is solved using the Numerov method for numerical integration.
Boundary Matching
The coefficients and are determined by matching the basis function and its derivative at the muffin-tin boundary. Using the Rayleigh expansion of plane waves:
the matching conditions give:
where is the muffin-tin radius and are spherical Bessel functions.
Generalized Eigenvalue Problem
The electronic structure problem becomes a generalized eigenvalue problem:
where:
- is the Hamiltonian matrix
- is the overlap matrix
- are the expansion coefficients
The overlap matrix is not the identity because the basis functions are not orthogonal (unlike pure plane waves).
Python Implementation
A simplified LAPW implementation using Numerov for the radial part:
import numpy as np
import scipy
from scipy.special import spherical_jn
import matplotlib.pyplot as plt
import matplotlib as mpl
def set_publication_style():
"""Set publication-quality matplotlib style."""
mpl.rcParams.update({
'font.family': 'serif',
'font.size': 12,
'axes.labelsize': 14,
'axes.titlesize': 16,
'axes.linewidth': 1.2,
'axes.labelpad': 8,
'axes.titlepad': 10,
'xtick.labelsize': 12,
'ytick.labelsize': 12,
'xtick.direction': 'in',
'ytick.direction': 'in',
'xtick.top': True,
'ytick.right': True,
'xtick.major.size': 6,
'ytick.major.size': 6,
'xtick.major.width': 1.2,
'ytick.major.width': 1.2,
'legend.fontsize': 12,
'legend.frameon': False,
'lines.linewidth': 2,
'lines.markersize': 6,
'figure.dpi': 100,
'savefig.dpi': 300,
'savefig.bbox': 'tight'
})
set_publication_style()
def Numerov(F, dx, f0=0.0, f1=1e-3):
"""
Numerov method for solving second-order ODEs.
Solves: d^2y/dx^2 = F(x) * y(x)
"""
Nmax = len(F)
dx = float(dx)
Solution = np.zeros(Nmax, dtype=float)
Solution[0] = f0
Solution[1] = f1
h2 = dx * dx
h12 = h2 / 12
w0 = (1 - h12 * F[0]) * Solution[0]
Fx = F[1]
w1 = (1 - h12 * Fx) * Solution[1]
Phi = Solution[1]
for i in range(2, Nmax):
w2 = 2 * w1 - w0 + h2 * Phi * Fx
w0 = w1
w1 = w2
Fx = F[i]
Phi = w2 / (1 - h12 * Fx)
Solution[i] = Phi
return Solution
def NumerovGen(F, U, dx, f0=0.0, f1=1e-3):
"""
Generalized Numerov method for inhomogeneous ODEs.
Solves: d^2y/dx^2 = F(x) * y(x) + U(x)
"""
Nmax = len(F)
dx = float(dx)
Solution = np.zeros(Nmax, dtype=float)
Solution[0] = f0
Solution[1] = f1
h2 = dx * dx
h12 = h2 / 12
w0 = Solution[0] * (1 - h12 * F[0]) - h12 * U[0]
w1 = Solution[1] * (1 - h12 * F[1]) - h12 * U[1]
Phi = Solution[1]
for i in range(2, Nmax):
Fx = F[i]
Ux = U[i]
w2 = 2 * w1 - w0 + h2 * (Phi * Fx + Ux)
w0 = w1
w1 = w2
Phi = (w2 + h12 * Ux) / (1 - h12 * Fx)
Solution[i] = Phi
return Solution
def CRHS(E, l, R, Veff):
"""
Compute right-hand side for radial Schrödinger equation.
Parameters:
E: Energy
l: Angular momentum quantum number
R: Radial coordinate array
Veff: Effective potential array
Returns:
RHS array for Numerov method
"""
N = len(R)
RHS = np.zeros(N, dtype=float)
for i in range(N):
# Radial Schrödinger equation: -1/r * d^2/dr^2 * r + l(l+1)/r^2 + V
# Rearranged for Numerov: d^2u/dr^2 = [2(-E + l(l+1)/(2r^2) + V)] * u
RHS[i] = 2 * (-E + 0.5 * l * (l + 1) / (R[i] * R[i]) + Veff[i])
return RHS
def ashcroft_potential(r, Z=1, rc=0.89, r1=0.4, e2=14.4):
"""
Ashcroft empty-core pseudopotential.
Parameters:
r: Radial distance array
Z: Valence charge
rc: Core radius
r1: Inner radius (flat region)
e2: e^2 / (4πε₀) in eV·Å
"""
A = -Z * e2 / rc
B = Z * e2 / rc**2
x2 = rc - r1
# Cubic interpolation coefficients
M = np.array([[x2**3, x2**2], [3*x2**2, 2*x2]])
a, b = np.linalg.solve(M, np.array([A, B]))
V = np.zeros_like(r)
for i, ri in enumerate(r):
if ri <= r1:
V[i] = 0.0
elif ri < rc:
x = ri - r1
V[i] = a * x**3 + b * x**2
else:
V[i] = -Z * e2 / ri
return V
# Example: Solve radial Schrödinger equation for l=0
rmt = 3.0 # Muffin-tin radius
E = 2.0 # Linearization energy
l = 0 # Angular momentum
Z = 1 # Atomic number
# Radial grid
R = np.linspace(1e-5, rmt, 1000, endpoint=True)
Veff = ashcroft_potential(R, Z=Z)
# Solve for u_l
crhs = CRHS(E, l, R, Veff)
crhs[0] = 0.0 # Boundary condition at origin
u_l = Numerov(crhs, (R[-1] - R[0]) / (len(R) - 1.0))
# Normalize: ∫ r^2 u_l^2 dr = 1
norm = np.sqrt(scipy.integrate.simpson(u_l * u_l, x=R))
u_l /= norm
# Solve for energy derivative u_dot
inhom = -2 * u_l
u_dot = NumerovGen(crhs, inhom, (R[-1] - R[0]) / (len(R) - 1.0))
# Orthogonalize u_dot with respect to u_l
alpha = scipy.integrate.simpson(u_l * u_dot, x=R)
u_dot -= alpha * u_l
# Normalize u_dot
norm_dot = np.sqrt(scipy.integrate.simpson(u_dot * u_dot, x=R))
u_dot /= norm_dot
# Plot results
fig, axes = plt.subplots(1, 2, figsize=(12, 5))
# Left: Potential and wavefunctions
axes[0].plot(R, Veff, 'k--', linewidth=2, label='Potential V(r)')
axes[0].plot(R, u_l, 'b-', linewidth=2, label='u_l(r)')
axes[0].plot(R, u_dot, 'r-', linewidth=2, label='u̇_l(r)')
axes[0].set_xlabel('r (Å)')
axes[0].set_ylabel('Amplitude')
axes[0].set_title('Radial Wavefunctions')
axes[0].legend()
axes[0].grid(True, alpha=0.3)
# Right: Probability density
axes[1].plot(R, R * u_l, 'b-', linewidth=2, label='r * u_l(r)')
axes[1].plot(R, R * u_dot, 'r-', linewidth=2, label='r * u̇_l(r)')
axes[1].set_xlabel('r (Å)')
axes[1].set_ylabel('r * u(r)')
axes[1].set_title('Radial Probability Density')
axes[1].legend()
axes[1].grid(True, alpha=0.3)
plt.tight_layout()
plt.savefig('figures/lapw_radial_wavefunctions.png', dpi=300, bbox_inches='tight')
plt.show()
print(f"Normalization check:")
print(f" ∫ r^2 u_l^2 dr = {scipy.integrate.simpson(u_l * u_l, x=R):.6f}")
print(f" ∫ r^2 u_dot^2 dr = {scipy.integrate.simpson(u_dot * u_dot, x=R):.6f}")
print(f" ∫ r^2 u_l * u_dot dr = {scipy.integrate.simpson(u_l * u_dot, x=R):.6f}")Visualization
The following plot shows the radial wavefunctions and potential:
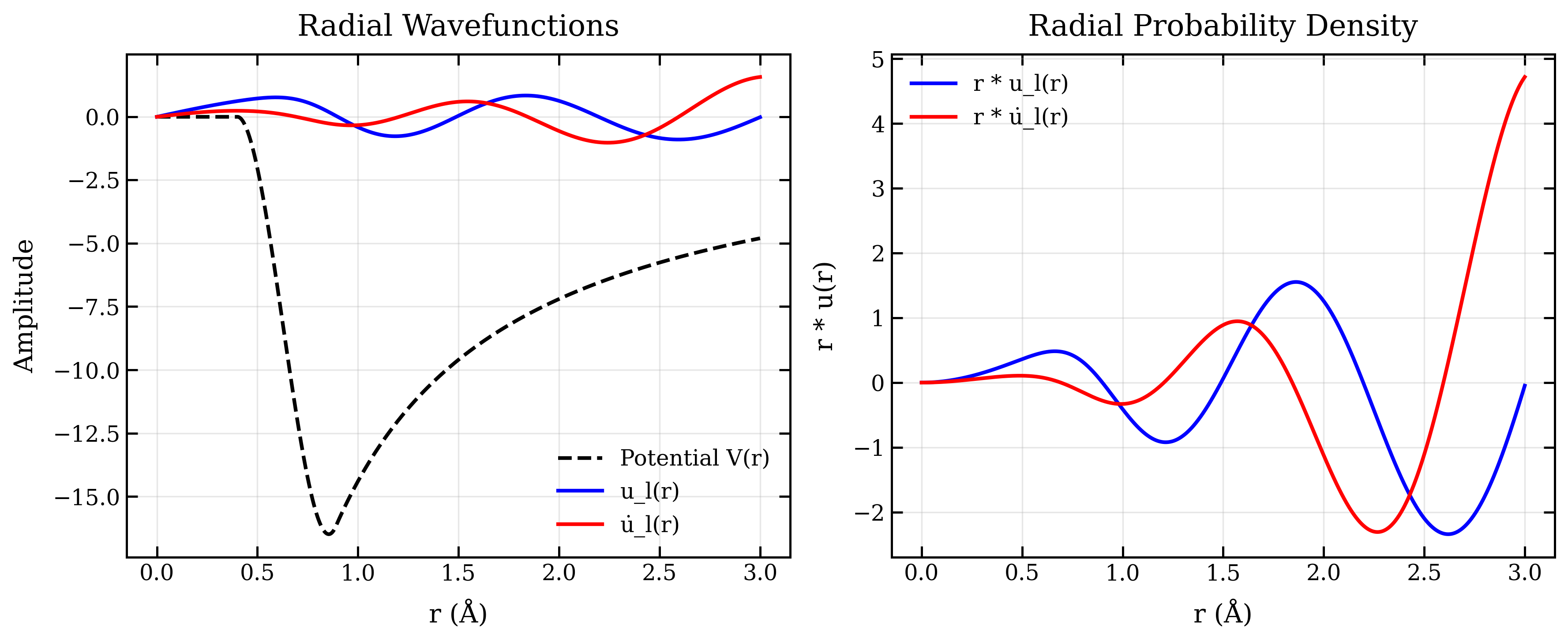
Key Features
- Piecewise basis functions adapted to the physics of each region
- Radial solutions computed using Numerov method
- Energy derivatives for linearization
- Boundary matching using spherical Bessel functions
- Generalized eigenvalue problem with overlap matrix
Advantages
The LAPW method offers several advantages:
- Accuracy: Handles both core and valence electrons accurately
- Efficiency: Fewer basis functions needed than pure plane waves
- Flexibility: Can handle strong potentials near nuclei
- All-electron: No pseudopotential approximation needed
Applications
LAPW is widely used in:
- Band structure calculations
- Density functional theory (DFT) for solids
- Magnetic materials
- Strongly correlated systems
- Transition metal compounds
Historical Context
The LAPW method was developed as an improvement over the Augmented Plane Wave (APW) method. The key innovation is the linearization, which allows the energy to vary while maintaining continuity at the muffin-tin boundary. This was first described by Koelling and Arbman in 1975.