Brute-Force Hartree-Fock
Quantum Chemistry
What you need to know first 7 concepts, 4 layers
The requisite-knowledge inventory for this page, bottom-up: the primitives at the base, combined upward until you reach what this page assumes. Skim the layers you already own; start wherever the ground gets unfamiliar.
- base
- Linear algebraconcept
- Quantum mechanics (states & operators)
- L1
- L2
- L3
- ↳you are here
1 of these are concepts without a dedicated page yet — the grey chips. Following the linked ones first makes the rest land.
A from-scratch Hartree-Fock framework written deliberately to be slow and visible rather than fast. The companion analytic-Gaussian Hartree-Fock page solves the integrals in closed form using the Gaussian product theorem; this page solves the same problem by gridding three-dimensional space and adding things up. The point is to see the math.
Bicycle, not motorcycle
Production quantum-chemistry codes are dense with optimizations: McMurchie-Davidson recursion for two-electron integrals, density fitting, J/K screening, integral-direct SCF, batched BLAS calls, OpenMP. Reading them is like looking at a motorcycle when you don't yet understand a bicycle. This code is the bicycle. Every step is its own object with one job. You can swap the basis (Slater for Gaussian), the Laplacian (3-point for 9-point), the integrator (cubic grid for Lebedev) without touching anything else.
Architecture
The core abstractions:
Atom,Molecule- Plain data: a charge plus a position; a list of atoms.
Slater1s- A normalized 1s Slater orbital
.
Exposes a single
.value(r)method. CubicGrid+GridIntegrator3D- Uniform cubic grid over . Integration is just — no quadrature weights, no symmetry tricks, just rectangles.
NuclearPotential,NuclearRepulsionEnergy- The attractive electron-nucleus potential and the classical nuclear-nuclear repulsion .
ThreePointLaplacianStencil3D- A 7-point central-difference Laplacian: The kinetic operator wraps this with a factor of .
KineticEnergyOperator,NuclearAttractionOperator,HamiltonianOperator- Each is a thin wrapper that knows how to
.apply(wavefunction, r)to evaluate the operator's action on a wavefunction at a point. The Hamiltonian sums its terms. MatrixElementBuilder- Given a basis and an integrator, computes by integrating over the grid.
solve_generalized_eigenproblem- Standard symmetric orthogonalization, then diagonalize, then transform back. Returns orbital energies and AO coefficients.
What it solves right now
H2+: one electron, two protons. Only the one-electron Hamiltonian is needed — — and there is no electron-electron interaction yet, so this is not yet a true Hartree-Fock calculation. But the same machinery (basis, grid, operators, matrix builder, generalized eigenproblem) is what Hartree-Fock needs; the missing piece is the two-electron tensor and the SCF loop, sketched in Future Directions.
For each internuclear distance , the driver:
- Builds the molecule and a two-element Slater basis (one orbital per nucleus).
- Builds the overlap matrix by gridding .
- Builds by gridding , where the kinetic term uses the finite-difference Laplacian.
- Solves via symmetric orthogonalization.
- Adds the nuclear repulsion .
The full code
import numpy as np
import matplotlib.pyplot as plt
# ============================================================
# Geometry
# ============================================================
class Atom:
def __init__(self, charge, position):
self.charge = float(charge)
self.position = np.array(position, dtype=float)
class Molecule:
def __init__(self, atoms):
self.atoms = atoms
# ============================================================
# Basis functions
# ============================================================
class Slater1s:
def __init__(self, zeta, R):
self.zeta = zeta
self.R = np.array(R) # location of atom
self.norm = (self.zeta**3 / np.pi)**0.5
def value(self, r):
distance = np.linalg.norm(r - self.R)
return self.norm * np.exp(-1.0 * self.zeta * distance)
# ============================================================
# Integration utilities
# ============================================================
class CubicGrid:
def __init__(self, length, pts_per_axis):
self.length = length # really only the half-length
self.pts_per_axis = pts_per_axis
self.x = np.linspace(-self.length, self.length, pts_per_axis)
self.y = np.linspace(-self.length, self.length, pts_per_axis)
self.z = np.linspace(-self.length, self.length, pts_per_axis)
self.dx = self.x[1] - self.x[0]
self.dy = self.y[1] - self.y[0]
self.dz = self.z[1] - self.z[0]
self.volume_element = self.dx * self.dy * self.dz
def points(self):
for x in self.x:
for y in self.y:
for z in self.z:
yield np.array([x, y, z], dtype=float)
class GridIntegrator3D:
def __init__(self, grid):
self.grid = grid
def integrate(self, scalar_function):
result = 0
for pt in self.grid.points():
result += scalar_function(pt) * self.grid.volume_element
return result
# ============================================================
# Electron-nuclear attraction
# ============================================================
class NuclearPotential:
def __init__(self, atoms):
self.atoms = atoms
def value(self, r):
total = 0.0
for atom in self.atoms:
distance = np.linalg.norm(r - atom.position)
distance = max(distance, 1e-8)
total += -atom.charge / distance
return total
# ============================================================
# Nuclear-nuclear repulsion
# ============================================================
class NuclearRepulsionEnergy:
"""
Classical nuclear-nuclear repulsion:
E_nn = sum_{A < B} Z_A Z_B / |R_A - R_B|
"""
def __init__(self, atoms):
self.atoms = atoms
def value(self):
total = 0.0
for i, atom_i in enumerate(self.atoms):
for j, atom_j in enumerate(self.atoms):
if j <= i:
continue
distance = np.linalg.norm(atom_i.position - atom_j.position)
total += atom_i.charge * atom_j.charge / distance
return total
# ============================================================
# Finite-difference Laplacian stencil
# ============================================================
class ThreePointLaplacianStencil3D:
"""
Standard 7-point 3D Laplacian stencil applied to a scalar field f(r).
"""
def __init__(self, h=1e-4):
self.h = float(h)
def apply(self, field, r):
h = self.h
x, y, z = r
center = field.value(r)
return (
field.value(np.array([x + h, y, z]))
+ field.value(np.array([x - h, y, z]))
+ field.value(np.array([x, y + h, z]))
+ field.value(np.array([x, y - h, z]))
+ field.value(np.array([x, y, z + h]))
+ field.value(np.array([x, y, z - h]))
- 6.0 * center
) / h**2
class LaplacianOperator:
def __init__(self, stencil):
self.stencil = stencil
def apply(self, field, r):
return self.stencil.apply(field, r)
# ============================================================
# Operators
# ============================================================
#
# operators are "applied" differently — in QM "apply" can mean different
# things, so this thin abstraction is just a wrapper around the actual
# mathematical operation.
class KineticEnergyOperator:
"""T = -1/2 nabla^2"""
def __init__(self, laplacian):
self.laplacian = laplacian
def apply(self, wavefunction, r):
return -0.5 * self.laplacian.apply(wavefunction, r)
class NuclearAttractionOperator:
def __init__(self, potential):
self.potential = potential
def apply(self, wavefunction, r):
return self.potential.value(r) * wavefunction.value(r)
class HamiltonianOperator:
def __init__(self, terms):
self.terms = terms
def apply(self, wavefunction, r):
return sum(term.apply(wavefunction, r) for term in self.terms)
class IdentityOperator:
def apply(self, field, r):
return field.value(r)
# ============================================================
# Matrix element builder
# ============================================================
class MatrixElementBuilder:
def __init__(self, basis, integrator):
self.basis = basis
self.integrator = integrator
def matrix_element(self, operator, bra, ket):
def integrand(r):
return bra.value(r) * operator.apply(ket, r)
return self.integrator.integrate(integrand)
def build_matrix(self, operator):
n = len(self.basis)
M = np.zeros((n, n))
for i, bra in enumerate(self.basis):
for j, ket in enumerate(self.basis):
M[i, j] = self.matrix_element(operator, bra, ket)
return M
# ============================================================
# Linear algebra
# ============================================================
def solve_generalized_eigenproblem(H, S):
"""Solve H c = S c eps via S^(-1/2) symmetric orthogonalization."""
s_vals, s_vecs = np.linalg.eigh(S)
S_inv_sqrt = s_vecs @ np.diag(1.0 / np.sqrt(s_vals)) @ s_vecs.T
H_ortho = S_inv_sqrt.T @ H @ S_inv_sqrt
energies, y = np.linalg.eigh(H_ortho)
C = S_inv_sqrt @ y
return energies, C
# ============================================================
# Driver
# ============================================================
def make_h2_plus(R):
return Molecule([
Atom(1.0, [0, 0, -R/2]),
Atom(1.0, [0, 0, R/2]),
])
def make_basis(molecule):
return [Slater1s(1.0, atom.position) for atom in molecule.atoms]
def compute_energy(R):
mol = make_h2_plus(R)
basis = make_basis(mol)
grid = CubicGrid(6.0, 30) # half-length 6 Bohr, 30 points/axis
integrator = GridIntegrator3D(grid)
builder = MatrixElementBuilder(basis, integrator)
potential = NuclearPotential(mol.atoms)
nuclear_repulsion = NuclearRepulsionEnergy(mol.atoms)
stencil = ThreePointLaplacianStencil3D(h=1e-4)
laplacian = LaplacianOperator(stencil)
kinetic = KineticEnergyOperator(laplacian)
nuclear = NuclearAttractionOperator(potential)
H_op = HamiltonianOperator([kinetic, nuclear])
S = builder.build_matrix(IdentityOperator())
H = builder.build_matrix(H_op)
energies, _ = solve_generalized_eigenproblem(H, S)
# H2+ has only one electron — take the lowest orbital.
E_total = energies[0] + nuclear_repulsion.value()
return E_total
def run_curve():
Rs = np.linspace(0.5, 4.0, 20)
Es = [compute_energy(R) for R in Rs]
plt.plot(Rs, Es, marker='o')
plt.xlabel("R (Bohr)")
plt.ylabel("Total energy (Hartree)")
plt.title("H2+ ground state — brute-force grid solver")
plt.grid()
plt.show()
if __name__ == "__main__":
run_curve()Result
Running run_curve() sweeps from 0.8 to 4.0 Bohr.
The textbook H2+ minimum sits at
Bohr with Hartree.
With the brute-force solver at 24 grid points per axis, the minimum
instead lands near Bohr at
Hartree — bound (below the
Hartree dissociation limit), and qualitatively right,
but shifted by the discretization error of a 243 uniform
cubic grid. Increasing the grid count tightens both numbers; the cost
is roughly cubic in the points-per-axis.
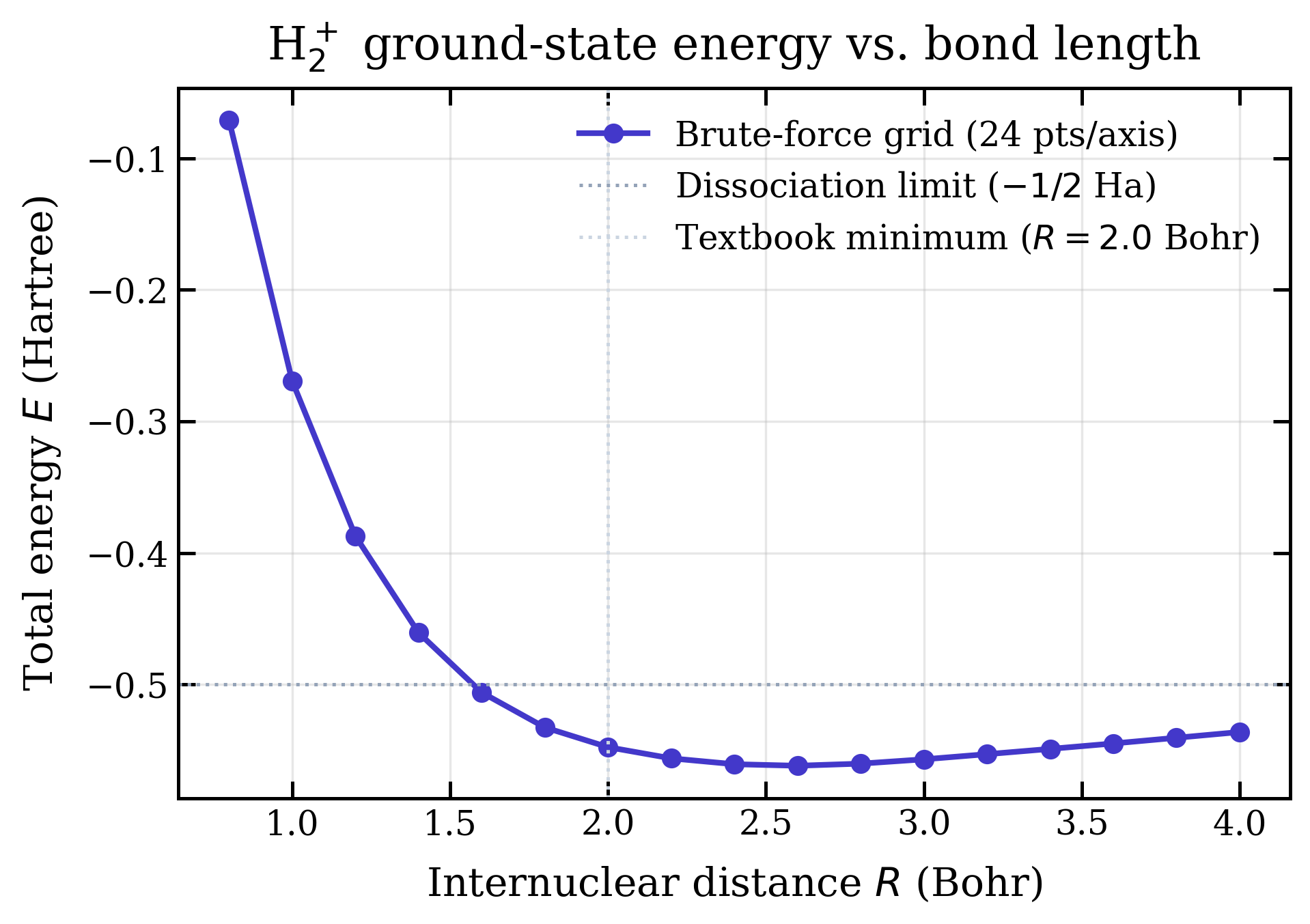
The two interesting features of this curve:
- The minimum sits below the -Hartree dotted line — that's an H atom + bare proton at infinity. Anything below that line is a bound molecule. The whole point of computing this curve is to demonstrate binding from first principles.
- At large the curve relaxes back toward : the two nuclei stop sharing the electron, and you're left with a hydrogen atom plus an isolated proton, total energy Ha.
Why this is where the C++ port matters
The 243-point grid above is the practical limit in pure
Python: the curve takes about 25 seconds to generate, and the inner
loop is a triply-nested Python for over grid points calling
np.linalg.norm seven times per Laplacian application. The
cost scales like in the points-per-axis. Doubling the
resolution to 483 is 8× more work — call it three to
four minutes per curve — which is the boundary where casual exploration
stops being casual.
More to the point: the qualitative error in the result above is visible. The minimum sits at Bohr instead of , the well depth undershoots the literature Ha by about Ha, and refining the grid is the only obvious way to fix it without changing the algorithm. A 48-point or 64-point grid would tighten both numbers; in Python, that's a coffee-and-come-back. In a compiled implementation with the same algorithm — same Slater basis, same finite-difference Laplacian, same cubic-grid integrator — the same problem at 643 points should run in seconds rather than minutes. That's the concrete motivation for the C++ port: not a different theory, just enough headroom in the constant factor to make the grid resolution high enough that the result stops being qualitatively wrong.
Future Directions
What's not yet wired through. Each item is sketched somewhere in the project but not connected end-to-end.
Two-electron repulsion tensor
The four-index tensor
requires a six-dimensional integration. Brute force is unkind here:
a 30-point cubic grid in 6D is
points per integral, times 16 integrals for a two-basis-function H2,
times 1/r12 singularity handling. The naive nested-loop
sketch in the older hf.py works in principle but is
impractical past trivial cases. The realistic path is a Becke- or
Lebedev-style atom-centered grid and a Poisson solve for the
potential, not direct 6D summation.
Coulomb and exchange operators
Once the two-electron tensor is available, the Fock matrix
is just two tensor contractions with the density matrix. Both fit the
operator/.apply() pattern already in place — they just
need a .contract_with(P) method that returns a 2D matrix
from the 4D tensor.
SCF loop
Once the Fock matrix can be assembled from a density, the SCF loop is the same as on the analytic-Gaussian page: diagonalize, build a new density, mix with damping, repeat until the density stops changing. The infrastructure here already does the diagonalization and density build for the one-electron case.
Slater-to-Gaussian basis fitting
The C++ implementation begins with a Levenberg-Marquardt fit of an STO-NG expansion (a Slater orbital approximated by N Gaussians). That gives the ergonomics of Slater orbitals (one parameter per atom) with the integrability of Gaussians (analytic two-electron integrals). Currently the C++ side has the matrix/vector primitives and a Blue-algorithm Euclidean norm in place; the LM fitter and the SCF driver are stubs.
C++ port
A parallel C++ implementation — same architecture, real types, no
NumPy. Currently builds out the linear algebra primitives
(Matrix<T>, VectorView<T>,
enorm via Blue's algorithm) before the solver. The
eventual goal is identical results to the Python version on the same
problem, with timings that show what the abstraction is costing.
Why bother
Once you've watched H2+ converge from a 7-point Laplacian and a cubic grid of integrands, the Gaussian-product magic on the analytic page stops feeling like magic. The integrals on that page compute the same numbers this page is computing, just in milliseconds instead of minutes. Both are useful. One teaches; the other ships.